Copyright © 2007-2016, Craig A. James
Content is available under GNU Free Documentation License 1.2
Source for this document: https://github.com/opensmiles/OpenSMILES
Contributors: Richard Apodaca, Noel O’Boyle, Andrew Dalke, John van Drie, Peter Ertl, Geoff Hutchison, Craig A. James, Greg Landrum, Chris Morley, Egon Willighagen, Hans De Winter, Tim Vandermeersch, John May
1. Introduction
1.1. Purpose
This document formally defines an open specification version of the SMILES language, a typographical line notation for specifying chemical structure. It is hosted under the banner of the Blue Obelisk project, with the intent to solicit contributions and comments from the entire computational chemistry community.
1.2. Motivation
SMILES was originally developed as a proprietary specification by
Daylight Chemical Information Systems
Since the introduction of SMILES in the late 1980’s, it has
become widely accepted as a defacto standard for exchange of molecular structures. Many independent
SMILES software packages have been written in C, C++, Java, Python, LISP, and probably even
FORTRAN.
At this point in the history of SMILES, it is appropriate for the chemistry community to develop a new, non-proprietary specification for the SMILES language. Daylight’s SMILES Theory Manual has long been the "gold standard" for the SMILES language, but as a proprietary specification, it limits the universal adoption of SMILES, and has no mechanism for contributions from the chemistry community. We salute Daylight for their past contributions, and the excellent SMILES documentation they provided free of charge for the past two decades.
1.3. Audience
This document is intended for developers designing or improving a SMILES parser or writer. Readers are expected to be acquainted with SMILES. Due to the formality of this document, it is not a good tutorial for those trying to learn SMILES. This document is written with precision as the primary goal; readability is secondary.
1.4. What is a Molecule? The Valence Model of Chemistry
Before defining the SMILES language, it is important to state the physical model on which it is based: the valence model of chemistry, which uses a mathematician’s graph to represent a molecule. In a chemical graph, the nodes are atoms, and the edges are semi-rigid bonds that can be single, double, or triple according to the rules of valence bond theory.
This simple mental model has little resemblance to the underlying quantum-mechanical reality of electrons, protons and neutrons, yet it has proved to be a remarkably useful approximation of how atoms behave in close proximity to one another. However, the valence model is an imperfect representation of molecular structure, and the SMILES language inherits these imperfections. Chemical bonds are often tautomeric, aromatic or otherwise fractional rather than neat integer multiples. Delocalized bonds, bond-centered bonds, hydrogen bonds and various other inter-atom forces that are well characterized by a quantum-mechanics description simply don’t fit into the valence model.
McLeod and Peters' quip captures the deficiencies of SMILES well: if you can’t build a molecule from a modeling kit, the deficiencies of SMILES and other connection-table formats become apparent.
2. Formal Grammar
2.1. Syntax versus Semantics
This SMILES specification is divided into two distinct parts: A syntactic specification specifies how the atoms, bonds, parentheses, digits and so forth are represented, and a semantic specification that describes how those symbols are interpreted as a sensible molecule. For example, the syntax specifies how ring closures are written, but the semantics require that they come in pairs. Likewise, the syntax specifies how atomic elements are written, but the semantics determines whether a particular ring system is actually aromatic.
For this specification, the syntax and semantics are explained separately; in practice, the syntax and semantics are usually mixed together in the code that implements a SMILES parser. This chapter is only concerned with syntax.
2.2. Grammar
| Section | Formal Grammar |
|---|---|
ATOMS |
|
atom ::= bracket_atom | aliphatic_organic | aromatic_organic | |
|
ORGANIC SUBSET ATOMS |
|
aliphatic_organic ::= |
|
aromatic_organic ::= |
|
BRACKET ATOMS |
|
bracket_atom ::= |
|
symbol ::= element_symbols | aromatic_symbols | |
|
isotope ::= NUMBER |
|
element_symbols ::= |
|
aromatic_symbols ::= |
|
CHIRALITY |
|
chiral ::= |
|
HYDROGENS |
|
hcount ::= |
|
CHARGES |
|
charge ::= |
|
ATOM CLASS |
|
class ::= |
|
BONDS AND CHAINS |
|
bond ::= |
|
ringbond ::= bond? DIGIT | bond? |
|
branched_atom ::= atom ringbond* branch* |
|
branch ::= |
|
chain ::= branched_atom | chain branched_atom | chain bond branched_atom | chain dot branched_atom |
|
dot ::= |
|
SMILES STRINGS |
|
smiles ::= terminator | chain terminator |
|
terminator ::= SPACE | TAB | LINEFEED | CARRIAGE_RETURN | END_OF_STRING |
|
3. Reading SMILES
3.1. Atoms
3.1.1. Atomic Symbol
An atom is represented by its atomic symbol, enclosed in square brackets, [].
The first character of the symbol is uppercase and the second (if any) is lowercase,
except that for aromatic atoms (see below), the first character is lowercase. There are
114 valid atomic symbols
, as defined by IUPAC (see also
Web Elements).
The symbol '*' is also accepted as a valid atomic symbol, and represents a "wildcard" or unknown atom.
Examples:
| SMILES | Atomic Symbol |
|---|---|
|
Uranium |
|
Lead |
|
Helium |
|
Unknown atom |
3.1.2. Hydrogens
Hydrogens inside of brackets are specified as Hn where n is a number such as H3. If no
Hn is specified, it is identical to H0. If H is
specified without a number, it is identical to H1. For example, [C] and
[CH0] are identical, and [CH] and [CH1] are identical.
Hydrogens that are specified in brackets with this notation have undefined isotope, no chirality, no other bound hydrogen, neutral charge, and an undefined atom class.
Examples:
| SMILES | Name | Comments |
|---|---|---|
|
methane |
|
|
hydrochloric acid |
|
|
hydrochloric acid |
A hydrogen atom cannot have a hydrogen count, for example, [HH1] is illegal. Hydrogens connected
to other hydrogens must be represented as explicit atoms in square brackets. For example molecular
hydrogen must be written as [H][H].
Question: are more than 9 hydrogens possible? Should they be supported?
3.1.3. Charge
Charge is specified by a +n or -n where n is a number; if the
number is missing, it means either +1 or -1 as appropriate.
For backwards compatibility, a general-purpose SMILES parser should
accept the symbols -- and ++ to mean charges of -2 and +2, but this
is a deprecated form and should be avoided.
Examples:
| SMILES | Name | Comments |
|---|---|---|
|
chloride anion |
|
|
hydroxyl anion |
|
|
hydroxyl anion |
|
|
copper cation |
|
|
copper cation |
|
An implementation is required to accept charges in the range -15 to +15.
3.1.4. Isotopes
Isotopic specification is placed inside the square brackets for an atom preceding the atomic symbol; for example:
| SMILES | Atomic Symbol |
|---|---|
|
methane |
|
deuterium ion |
|
Uranium 238 atom |
An isotope is interpreted as a number, so that [2H],
[02H] and [002H] all mean deuterium. If the isotope field
is not specified then the atom is assumed to have the naturally-occurring isotopic ratios. The
isotope value 0 also indicates an isotope of zero, that is
[0S] is not the same as [S].
There is no requirement that the isotope is a genuine isotope of the element. Thus,
[36Cl] is allowed even though 35Cl and
37Cl are the actual known stable isotopes of chlorine.
A general-purpose SMILES parser must accept at least three digits for the isotope and values from 0 to 999.
3.1.5. Organic Subset
A special subset of elements called the "organic subset" of B, C, N, O, P, S, F, Cl, Br, I, and * (the "wildcard" atom) can be written using the only the atomic symbol (that is, without the square brackets, H-count, etc.). An atom is specified this way has the following properties:
-
"implicit hydrogens" are added such that valence of the atom is in the lowest normal state for that element
-
the atom’s charge is zero
-
the atom has no isotopic specification
-
the atom has no chiral specification
The implicit hydrogen count is determined by summing the bond orders of the bonds connected to the atom. If that sum is equal to a known valence for the element or is greater than any known valence then the implicit hydrogen count is 0. Otherwise the implicit hydrogen count is the difference between that sum and the next highest known valence.
The "normal valence" for these elements is defined as:
| Element | Valence |
|---|---|
B |
3 |
C |
4 |
N |
3 or 5 |
O |
2 |
P |
3 or 5 |
S |
2, 4 or 6 |
halogens |
1 |
* |
unspecified |
Examples:
| SMILES | Name |
|---|---|
|
methane |
|
ammonia |
|
hydrochloric acid |
Note: The remaining atom properties, chirality and ring-closures, are discussed in later sections.
3.1.6. The Wildcard '*' Atomic Symbol
The '*' atom represents an atom whose atomic number is unknown or unspecified. If it occurs
inside brackets, it can have its isotope, chirality, hydrogen count and charge specified. If it
occurs outside of brackets, it has no assumed isotope, a mass of zero, unspecified chirality, a
hydrogen count of zero, and a charge of zero.
| SMILES | Name |
|---|---|
|
ortho-substituted phenol |
The '*' atom does not have any specific electronic properties or
valence. If specified outside of square brackets, it takes on the valence
implied by its bonds. If it is inside square brackets, it takes on the
valence implied by its bonds, hydrogens and/or charge.
A '*' atom can be part of an aromatic ring. When deducing the
aromaticity of a ring system, the ring system is considered aromatic if
there is an element which could replace the '*' and make the ring system
meet the aromaticity rules (see Aromaticity, below).
3.1.7. Atom Class
An "atom class" is an arbitrary integer, a number that has no chemical meaning. It is used by applications to mark atoms in ways that are meaningful only to the application. Multiple atoms may be labeled with the same atom class.
The atom class is specified after all other properties in square brackets. For example:
| SMILES | Name |
|---|---|
|
methane, atom’s class is 2 |
If the atom class is not specified then the atom class is zero.
The atom class is interpreted as a number, so both [CH2:5]
and [NH4+:005] have an atom class of 5.
3.2. Bonds
Atoms that are adjacent in a SMILES string are assumed to be joined by a single or aromatic bond (see Aromaticity). For example:
| SMILES | Name |
|---|---|
|
ethane |
|
ethanol |
|
n-butylamine |
|
n-butylamine |
Double, triple and quadruple bonds are represented by '=', '#', and '$' respectively:
| SMILES | Name |
|---|---|
|
ethene |
|
hydrogen cyanide |
|
2-butyne |
|
propanol |
|
octachlorodirhenate (III) |
A single bond can be explicitely represented with '-', but it is rarely
necessary.
| SMILES | |
|---|---|
|
same as: |
|
same as: |
|
same as: |
Note: The remaining bond symbols, ':\/', are discussed in
later sections.
3.3. Branches
An atom with three or more bonds is called a branched atom, and is represented using parentheses.
To maintain a valid structure, any nested branch (a branch within another branch) must be preceded by a valid atom, meaning that you cannot start a nested branch until there is a clearly defined atom in the parent branch.
For example, C(C(C)) is valid, as each branch has an atom before moving to a nested branch. However, C((C)C) is not valid, because the nested branch starts without a proper atom preceding it.
| Depiction | SMILES | Name |
|---|---|---|
|
|
2-ethyl-1-butanol |
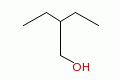
Branches can be nested or "stacked" to any depth:
| Depiction | SMILES | Name |
|---|---|---|
|
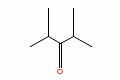
|
2,4-dimethyl-3-pentanone |
pic here |
|
2-propyl-3-isopropyl-1-propanol |
|
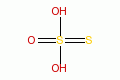
|
thiosulfate |
The SMILES branch/chain rules allow nested parenthetical expressions (branches) to an arbitrary depth. For example, the following SMILES, though peculiar, is legal:
| SMILES | Formula |
|---|---|
|
C22H46 |
3.4. Rings
In a SMILES string such as "C1CCCCC1", the first occurrence of a ring-closure number (an "rnum") creates an "open bond" to the atom that precedes the ring-closure number (the "rnum"). When that same rnum is encountered later in the string, a bond is made between the two atoms, which typically forms a cyclic structure.
| Depiction | SMILES | Name |
|---|---|---|
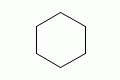
|
|
cyclohexane |
|
|
perhydroisoquinoline |
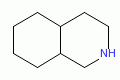
If a bond symbol is present between the atom and rnum, it can be present on either or both bonded atoms. However, if it appears on both bonded atoms, the two bond symbols must be the same.
| Depiction | SMILES | Name |
|---|---|---|
|
|
cyclohexene |
|
cyclohexene (preferred form) |
|
|
cyclohexene |
|
|
invalid |
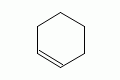
Ring closures must be matched pairs in a SMILES string, for example, C1CCC
is not a valid SMILES.
It is permissible to re-use ring-closure numbers. Once a particular number has been encountered twice, that number is available again for subsequent ring closures.
| Depiction | SMILES | Name | Comment |
|---|---|---|---|
|
|
bicyclohexyl |
both SMILES are valid |
|
bicyclohexyl |
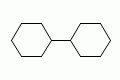
Note that the ring number zero is valid, for example cyclohexane can be
written C0CCCCC0.
Two-digit ring numbers are permitted, but must be preceded by the percent
'%' symbol, such as C%25CCCCC%25 for cyclohexane. Three-digit numbers and
larger are never permitted. However, note that three digits are not invalid; for
example, C%123 is the same as C3%12, that is, an atom with two rnum
specifications.
The digit(s) representing a ring-closure are interpreted as a number, not a
symbol, and two rnums match if their numbers match. Thus, C1CCCCC%01 is a
valid SMILES and is the same as C1CCCCC1. Likewise, C%00CCCCC%00 is a
valid SMILES.
A single atom can have several ring-closure numbers, such as this spiro atom:
| Depiction | SMILES | Name |
|---|---|---|
|
|
spiro[5.5]undecane |
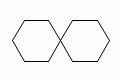
Two atoms cannot be joined by more than one bond, and an atom cannot be bonded to itself. For example, the following are not allowed:
| SMILES | Comments |
|---|---|
|
illegal, two bonds between one pair of atoms |
|
illegal, two bonds between one pair of atoms |
|
illegal, atom bonded to itself |
3.5. Aromaticity
3.5.1. The Meaning of "Aromaticity" in SMILES
"Aromaticity" in SMILES is primarily for cheminformatics purposes. In a cheminformatics system, we’d like to have a single representation for each molecule. The Kekule form masks the inherent uniformity of the bonds in an aromatic ring. SMILES uses a simplified definition of aromaticity that facilitates substructure and exact-structure searches, as well as Normalization and Canonicalization of SMILES.
The definition of "aromaticity" in SMILES is not intended to imply anything about the physical or chemical properties of a substance. In many or most cases, the SMILES definition of aromaticity will match the chemist’s notion of what is aromatic, but in some cases it will not.
3.5.2. Kekule and Aromatic Representations
Aromaticity can be represented in one of two ways in a SMILES.
-
In the Kekule form, using alternating single and double bonds, with uppercase symbols for the atoms.
-
An atomic symbol that begins with a lowercase letter is an aromatic atom, such as
'c'for aromatic carbon. When aromatic symbols are used, no bond symbols are needed.
A lowercase aromatic symbol is defined as an atom in the sp2 configuration in an aromatic or anti-aromatic ring system. For example:
| Depiction | SMILES | Name |
|---|---|---|
|
|
benzene |
|
||
|
|
indane |
|
||
|
|
furan |
|
||
|
|
cyclobutadiene |
|
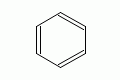
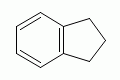
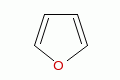

The Kekule form is always acceptable for SMILES input. For output, the aromatic form (using lowercase letters) is preferred. The lowercase symbols eliminate the arbitrary choice of how to assign the single and double bonds, and provide a normalized form that more accurately reflects the electronic configuration.
3.5.3. Extended Hueckel’s Rule
THIS SECTION IS UNDER MAJOR REVISION, AND AT THIS POINT IS ONLY FOR DISCUSSION PURPOSES.
This proposed section is an attempt to simplify the rule-based system by enumerating all atom/bond configurations that are known to participate in aromatic systems.
A single, isolated ring that meets the following criteria is aromatic:
-
All atoms must be sp2 hybridized.
-
The number of available "shared" π electrons must equal 4N+2 where N ≥ 0 (Huckel’s rule).
Each element that can participate in an aromatic ring is defined to have the following number of π electrons:
| Configuration | π Electrons | Example | Comment |
|---|---|---|---|
|
0 |
|
OpenSMILES extension |
|
1 |
|
OpenSMILES extension |
|
2 |
|
|
|
0 |
|
|
|
0 |
|
|
|
1 |
|
|
|
1 |
|
|
|
1 |
|
|
|
2 |
|
|
|
2 |
|
|
|
1 |
|
|
|
1 |
|
|
|
1 |
|
Non-oxide contributes 2 in Daylight toolkit |
|
2 |
|
|
|
2 |
|
|
|
1 |
|
|
|
1 |
|
|
|
1 |
|
Non-oxide contributes 2 in Daylight toolkit |
|
2 |
|
|
|
1 |
|
OpenSMILES extension |
|
1 |
|
OpenSMILES extension |
|
2 |
|
|
|
1 |
|
|
|
2 |
|
|
|
1 |
|
|
|
2 |
|
Possibly chiral |
|
2 |
|
Possibly chiral, OpenSMILES extension |
|
2 |
|
|
|
1 |
|
|
|
2 |
|
Possibly chiral |
|
2 |
|
Possibly chiral, OpenSMILES extension |






























































3.5.4. Aromaticity Algorithm
In an aromatic system, all of the aromatic atoms must be sp2 hybridized, and the number of π electrons must meet Huckel’s 4n+2 criterion When parsing a SMILES, a parser must note the aromatic designation of each atom on input, then when the parsing is complete, the SMILES software must verify that electrons can be assigned without violating the valence rules, consistent with the sp2 markings, the specified or implied hydrogens, external bonds, and charges on the atoms.
The aromatic-bond symbol ':' can be used between aromatic atoms, but it is never necessary; a
bond between two aromatic atoms is assumed to be aromatic unless it is explicitly represented as a
single bond '-'. However, a single bond (nonaromatic bond) between two aromatic atoms must
be explicitly represented. For example:
| Depiction | SMILES | Name |
|---|---|---|
|
|
biphenyl |
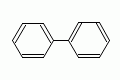
Note: Some SMILES parsers interpret a lowercase letter as sp2 anywhere it appears;
for example, CccccC would be interpreted as CC=CC=CC.
The OpenSMILES specification does not allow this interpretation unless
nonstandard parsing is explicitely allowed by the user.
3.6. More about Hydrogen
Hydrogens in a SMILES can be represented in three different ways:
| Method | SMILES | Name | Comments |
|---|---|---|---|
implicit hydrogen |
|
methane |
h-count deduced from normal valence (4) |
atom property |
|
methane |
h-count specified for heavy atom |
explicit hydrogen |
|
methane |
hydrogens represented as normal atoms |
All three forms are equivalent. However, some situations require that one form must be used:
-
Implicit hydrogen count may only be used for elements of the organic elements subset.
-
Any atom that is specified with square brackets must have its attached hydrogens explicitly represented, either as a hydrogen count or as normal atoms.
A hydrogen that meets one of the following criteria must be represented as an explicit atom:
-
hydrogens with charge (
[H+]) -
a hydrogen connected to another hydrogen (such as molecular hydrogen,
[H][H]) -
hydrogens with more than one bond (bridging hydrogens)
-
Deuterium
[2H]and tritium[3H]
It is permissible to use a mixture of an atom h-count and explicit hydrogen. In such a case, the atom’s hydrogen count is the sum of the atomic h-count property and the number of attached hydrogens. For example:
| SMILES | Name |
|---|---|
|
methane |
|
methane |
|
deuteroethane |
3.7. Disconnected Structures
The dot '.' symbol (also called a "dot bond") is legal most places where
a bond symbol would occur, but indicates that the atoms are not
bonded. The most common use of the dot-bond symbol is to represent
disconnected and ionic compounds.
| Depiction | SMILES | Name |
|---|---|---|
|
|
sodium chloride |
|
|
phenol, 2-amino ethanol |
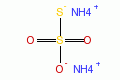
|
|
diammonium thiosulfate |
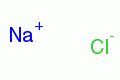
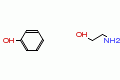
The dot can appear most places that a bond symbol is allowed, for example, the phenol example above can also be written:
| Depiction | SMILES | Name |
|---|---|---|
|
|
phenol, 2-amino ethanol |
|
phenal, 2-amino ethanol |
The second example above is an odd, but legal, use of parentheses and the dot bond, since the syntax allows a dot most places a regular bond could appear (the exception is that a dot can’t appear before a ring-closure digit).
Although dot-bonds are commonly used to represent compounds with disconnected parts, a dot-bond does not in itself mean that there are disconnected parts in the compound. See the following section regarding ring digits for some examples that illustrate this.
The dot bond cannot be used in front of a ring-closure digit. For example, C.1CCCCC.1 is illegal.
3.7.1. Other Uses of Ring Numbers and Dot Bond
A ring-number specifications ("rnum") is most commonly used to specify a ring-closure bond, but
when used with the '.' dot-bond symbol, it can also specify a non-ring bond. Two rnums in a SMILES
mean that the two atoms that precede the rnums are bonded. A dot-bond '.' means that the atoms to
which it is adjacent in the SMILES string are not bonded to each other. By combining these
two constructs, one can "piece together" fragments of SMILES into a whole molecule. The following
SMILES illustrate this:
| SMILES/Depiction | Fragment SMILES | Name |
|---|---|---|
|
|
ethane |
|
|
propane |
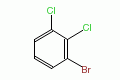
|
|
1-bromo-2,3-dichlorobenzene |
This feature of SMILES provides a convenient method of enumerating the molecules of a combinatorial library using string concatenation.
3.8. Stereochemistry
3.8.1. Scope of Stereochemistry in SMILES
A SMILES string can specify the cis/trans configuration around a double bond, and can specify the chiral configuration of specific atoms in a molecule.
SMILES strings do not represent all types of stereochemistry. Examples of stereochemistry that cannot be encoded into a SMILES include:
-
Gross conformational left or right handedness such as helices
-
Mechanical interferences, such as rotatable bonds that are constrained by mechanical interferences
-
Gross conformational stereochemistry such as the shape of a protein after folding
3.8.2. Tetrahedral Centers
SMILES uses an atom-centered chirality specification, in which the atom’s left-to-right order in the SMILES string itself is used as the basis for the chirality marking.
| Tetrahedral Chirality | |
|---|---|
look from N towards C (chiral center) |
list the neighbors anticlockwise |
|
|
…or clockwise |
|
|
|
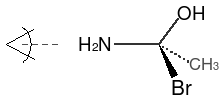
For the structure above, starting with the nitrogen atom, one "looks" toward the chiral
center. The remaining three neighbor atoms are written by listing them in anticlockwise order using the '@'
chiral property on the atom, or in clockwise order using the '@@' chiral property, as illustrated
above. The '@' symbol is a "visual mnemonic" in that the spiral around the character goes in the
anticlockwise direction, and means "anticlockwise" in the SMILES string (thus, '@@' can be thought of
as anti-anti-clockwise).
A chiral center can be written starting anywhere in the SMILES string, and the choice of whether to list the remaining neighbor in clockwise or anticlockwise order is also arbitrary. The following SMILES are all equivalent and all specify the exact same chiral center illustrated above:
| Equivalent SMILES | |
|---|---|
|
|
|
|
|
|
|
|
|
|
One exception to the atom order is when these atoms are bonded to the chiral center via a ring bond. In these cases, it is to order of the bonds to these atoms that should be considered. The two SMILES below are equivalent:
| Equivalent SMILES | |
|---|---|
|
|
If one of the neighbor atoms is a hydrogen and is represented as an atomic property of the
chiral center (rather than explicitly as [H]), then it is considered to be the first atom in the
clockwise or anticlockwise accounting. For example, if we replaced the bromine in the illustration
above with a hydrogen atom, its SMILES would be:
| Implicit Hydrogen |
|---|
|
3.8.3. Cis/Trans configuration of Double Bonds
The configuration of atoms around double bonds is specified by the bond symbols '/' and '\'.
These symbols always come in pairs, and indicate cis or trans with a visual "same side" or
"opposite side" concept. That is:
| Depiction | SMILES | Name |
|---|---|---|
|
|
trans-difluoroethane (both SMILES are equivalent) |
|
||
|
|
cis-difluoroethane (both SMILES are equivalent) |
|
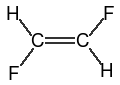
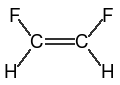
The "visual interpretation" of the '/' and '\' symbol is that they are thought of as bonds that
"point" above or below the alkene bond. That is, F/C=C/Br means "The F is below the first carbon,
and the Br is above the second carbon," leading to the interpretation of a trans configuration.
This notation can be confusing when parentheses follow one of the alkene carbons:
| SMILES | Name |
|---|---|
|
trans-difluoroethane |
|
|
|
cis-difluoroethane |
|
The "visual interpretation" of the "up-ness" or "down-ness" of each single bond is relative to the carbon atom, not the double bond, so the sense of the symbol changes when the fluorine atom moved from the left to the right side of the alkene carbon atom.
Note: This point was not well documented in earlier SMILES specifications, and several SMILES
interpreters are known to interpret the '/' and '\' symbols incorrectly.
A SMILES with conflicting up/down specifications is invalid:
| SMILES | Comment |
|---|---|
|
Invalid SMILES: Both the methyl and fluorine are "down" relative to the first allenal carbon |
It is permissible, but not required, that every atom attached to a double bond be marked. As long as at least two neighbor atoms, one on each end of the double bond, is marked, the "up-ness" or "down-ness" of the unmarked neighbors can be deduced.
| SMILES | Comment |
|---|---|
|
trans-difluoro configuration, position of methyl is implied |
Extended cis and trans configurations can be specified for conjugated allenes with an odd number of double bonds:
| SMILES | Name |
|---|---|
|
trans-difluorobutatriene |
|
cis-difluorobutatriene |
3.8.4. Tetrahedral Allene-like Systems
Extended tetrahedral configurations can be specified for conjugated allenes with an even number
of double bonds. The normal tetrahedral rules using '@' and '@@' apply, but the "neighbor" atoms
to which the chirality refers are at the ends of the allene system. For example:
| Depiction | SMILES |
|---|---|
|
|
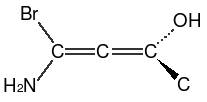
To determine the correct clockwise or anticlockwise specification, the allene is conceptually "collapsed" into a single tetrahedral chiral center, and the resulting chirality is marked as a property of the center atom of the extended allene system.
3.8.5. Square Planar Centers
There are three tags to represent square planar stereochemistry: @SP1, @SP2
and @SP3. Since there is no way to determine to what chirality class an atom
belongs based on the SMILES alone, the SP class is not the default class for
tetravalent stereocenters. Therefore are the shorthand notations (@, @@) not
equivalent to @SP1 and @SP2. That is, the full specification must be there
(@SP followed by 1, 2 or 3). The square planar also differs from the other
chiral primitives in that it does not use the notion of (anti-)clockwise.
Instead, each primitive represents a shape that is formed by drawing a line
starting from the atom that is first in the SMILES pattern to the next until
the end atom is reached. This may result in 3 possible shaped which are
referred to by a character with identical shape: 'U' for @SP1, '4' for @SP2 and
'Z' for @SP3. The graphical from of these shapes is illustrated in the image
below.
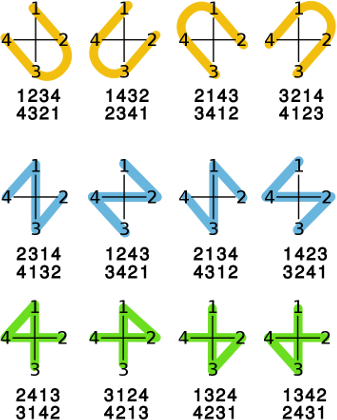
Background:
Also note that each shape starts and ends at specific positions. Both U and Z start from atoms that are successors or predecessors when arranging the atoms in the plane in anti-clockwise or clockwise order. The start and end atoms for the Z shape are never adjacent in such an ordering. For each shape there are 4 possible ways to start (and end) drawing the line. Also, for all the drawn lines, the start and end point can be exchanged. Thus 3 shapes, 4 ways to start/end and 2 ways to order the atoms for a shape results in 3 * 4 * 2 or 24 combinations. This is the same as the number of permutations that can be made with 4 numbers (i.e. P(n) = n!). This allows for canonical SMILES writers to use any ordering to output the atoms.
3.8.6. Trigonal Bipyramidal Centers
The chiral atom’s neighbors are labeled a, b, c, d, and e in the order that they
are parsed. For example, for S[As@@](F)(Cl)(Br)N S corresponds to a, F to b, Cl
to c, Br to d and N to e. This order is the unit permutation, represented as the
ordered set (a, b, c, d, e). In the simplest case @TB1 viewing from a towards e,
(b, c, d) are anti-clockwise (@). Likewise, @TB2 is specified as viewing from a
towards e, (b, c, d) are ordered clockwise (@@). The remaining TB’s permute the
axis as indicated in the table below. A final example, for @TB6 the viewing axis is from
a towards c and (b, d, e) are clockwise (@@).
| Viewing Axis | TB Number | Order | |
|---|---|---|---|
From |
Towards |
||
|
|
TB1 |
@ |
TB2 |
@@ |
||
|
|
TB3 |
@ |
TB4 |
@@ |
||
|
|
TB5 |
@ |
TB6 |
@@ |
||
|
|
TB7 |
@ |
TB8 |
@@ |
||
|
|
TB9 |
@ |
TB11 |
@@ |
||
|
|
TB10 |
@ |
TB12 |
@@ |
||
|
|
TB13 |
@ |
TB14 |
@@ |
||
|
|
TB15 |
@ |
TB20 |
@@ |
||
|
|
TB16 |
@ |
TB19 |
@@ |
||
|
|
TB17 |
@ |
TB18 |
@@ |
||
The following SMILES are all equivalent:
| Equivalent SMILES | |
|---|---|
|
|
|
|
|
|
A tool like Daylight’s depict match can help debugging
Background:
The trigonal Bipyramidal chirality is considerably more complex than any of the previous classes since the chiral atom has an extra neighbor. This increases the number of combinations to order the neighbors in a SMILES string from 24 to 120. Since every order of the atoms should be representable by a SMILES string, the 20 TB primitives suffice for this. In the trigonal bipyramidal geometry, 3 atoms lie in a plane and the remaining 2 atoms are perpendicular to this plane and are on the opposite sides of the plane forming an axis. The anti-clockwise and clockwise refers to the order of the 3 plane atoms when viewing along the axis in the specified direction. Unlike tetrahedral geometry, reordering the 3 atoms does not require that the axis be changed. Given an order of the axis atoms the 3 plane atoms are ordered either anti-clockwise or clockwise. Although there are P(3) = 3! or 6 possible permutations of 3 numbers, exchanging a pair inverts the parity and the 6 permutations are therefore divided in two groups (@, @@) containing 3 permutations each. Because there are now two atoms that determine the viewing direction along the axis, these atoms too can be in any of the 5 positions in a permutation. Given the atoms as the set {a, b, c, d, e}, there are C(5, 2) = 20 possible combinations of 5 things taken 2 at a time. However, the use of the @ and @@ symbols halve this to 10. These 10 combinations are the ordered sets (a, e), (a, d) (a, c), (a, b), (b, e), (b, d), (b, c), (c, e), (c, d) and (d, e). Each of these pairs correspond to an TB primitive.
3.8.7. Octahedral Centers
For 6 atoms, the unit permutation is (a, b, c ,d ,e ,f). @OH1 means when viewing
from a towards f, (b, c, d, e) are ordered anti-clockwise (@). @OH2 uses the same
axis but the 4 intermediate atoms are ordered clockwise. The interpretation of the 28
remaining numbers is more complex though. The concept of shapes (see square planar
stereochemistry) to describe the orientation of 4 atoms in a plane is reused. However,
this time these shapes also have a clockwise or anti-clockwise winding. For the U shape,
this is trivial since it means that the 4 atoms are listed clockwise or anti-clockwise.
For the Z shape, the connection between the first two atoms determines the winding.
Finally, for the 4 shape, the connection between the second and third atom determines
the winding. The table below lists the shapes, axes and orders.
| Shape | Viewing Axis | OH Number | Order | |
|---|---|---|---|---|
From |
Towards |
|||
|
|
|
OH1 |
@ |
OH2 |
@@ |
|||
|
|
OH3 |
@ |
|
OH16 |
@@ |
|||
|
|
OH6 |
@ |
|
OH18 |
@@ |
|||
|
|
OH19 |
@ |
|
OH24 |
@@ |
|||
|
|
OH25 |
@ |
|
OH30 |
@@ |
|||
|
|
|
OH4 |
@ |
OH14 |
@@ |
|||
|
|
OH5 |
@ |
|
OH15 |
@@ |
|||
|
|
OH7 |
@ |
|
OH17 |
@@ |
|||
|
|
OH20 |
@ |
|
OH23 |
@@ |
|||
|
|
OH26 |
@ |
|
OH29 |
@@ |
|||
|
|
|
OH10 |
@ |
OH8 |
@@ |
|||
|
|
OH11 |
@ |
|
OH9 |
@@ |
|||
|
|
OH13 |
@ |
|
OH12 |
@@ |
|||
|
|
OH22 |
@ |
|
OH21 |
@@ |
|||
|
|
OH28 |
@ |
|
OH27 |
@@ |
|||
The following SMILES are all equivalent:
| Equivalent SMILES | |
|---|---|
|
|
|
|
|
|
|
|
Background:
Octahedral stereochemistry is even more complicated since there is yet another extra neighboring atom. This raises the number of permutations to P(6) = 720. There are three axis that can be chosen and the orientation of the remaining 4 atoms has to be described. To describe these 4 atoms, P(4) = 24 permutations are used together with a shape. An axis always starts from the first neighbor atom and can end at any of the other neighbor atoms giving rise to 5 axis. As a result, each OH number encodes the axis positions, a shape and an order. Since all 3 axis can be placed in this positions, the start/end can be exchanged and each shape can start from any of the 4 atoms, each number represents 3 * 2 * 4 = 24 of the 720 permutations. Finally, 24 * 30 = 720 so all permutations can be used to write a canonical SMILES.
3.8.8. Partial Stereochemistry
SMILES allows partial stereochemical specifications. It is permissible for some chiral centers or double bonds to have stereochemical markings in the SMILES, while others in the same SMILES string do not. For example:
| SMILES | Comment |
|---|---|
|
completely specified |
|
partially specified |
|
partially specified |
3.8.9. Other Chiral Configurations
The SMILES language supports a number of atom-centered chiral configurations:
| SMILES | Configuration |
|---|---|
|
Tetrahedral |
|
Allenal |
|
Square Planar |
|
Trigonal Bipyramidal |
|
Octahedral |
The shorthand notations '@' and '@@' correspond to anti-clockwise and
clockwise tetrahedral chirality, and are the same a '@TH1' and
'@TH2', respectively. Likewise, in an allenal configuration, the shorthand
notations '@' and '@@' correspond to '@AL1' and '@AL2', respectively.
Very few SMILES systems actually implement the rules for SP, TB or OH chirality.
3.9. Parsing Termination
A SMILES string is terminated by a whitespace terminator character (space, tab, newline, carriage-return), or by the end of the string.
Other data or information, such as a name, properties, registration number, etc., may follow the SMILES on a line after the whitespace character. SMILES parsers will ignore this data, although applications that use the SMILES parser will often make use of it.
3.10. Programming Practices
OpenSMILES is designed to facilitate exchange of chemical information. To achieve that goal, it SMILES parsers should impose as few limits as possible on the language.
There is no formal limit to the length of a SMILES string; SMILES of over 1 million characters have been assembled for various purposes. There is no requirement that a SMILES parser must be able to parse these exceptionally long SMILES, but as a guideline, all implementations of SMILES parsers should, at a minimum, accept and correctly parse SMILES strings of 100,000 characters. If a SMILES parser encounters a string that is too long to parse, it should generate a relevant error message.
A SMILES parser should accept at least four digits for the atom class, and the values 0 to 9999.
There is no formal limit to the number of rings a molecule can contain. There are only 100 ring-closure numbers, but since numbers can be reused, a molecule can potentially have more than 100 rings. SMILES parsers should accept and correctly parse molecules with at least 1000 rings; it is preferable to place no limits on the number of rings a molecule can contain.
Branches (parentheses) can be nested to an arbitrary depth. Some SMILES strings in standard databases contain over 30 levels of branches, and much deeper nesting is possible. A general purpose parser must handle at least 100 levels; it is preferable to place no limits on nesting depth for parentheses.
There is no formal limit on the number of bonds an atom can have. SMILES parsers should allow at least ten bonds to each atom; it is preferable to place no limits on the number of bonds to each atom.
There is no limit to the number of "dot-disconnected" fragments in a SMILES. A SMILES of 100,000 atoms could in principle contain no bonds at all; SMILES parsers should place no limits on the number of fragments allowed (except that it is limited to the number of atoms the parser can manage).
Programmers are strongly encouraged to provide detailed and clear error messages. If possible, the error message should show exactly which character or "phrase" of the SMILES string triggered the error message.
4. Writing SMILES: Normalizations
4.1. What is Normalization?
A wide variety of SMILES strings are acceptable as input. For example, all of the following represent ethanol:
| SMILES | Name |
|---|---|
|
ethanol |
|
ethanol |
|
ethanol |
|
ethanol |
|
ethanol |
However, it is desirable to write SMILES in more standard forms; the first two forms above are preferred by most chemists, and require fewer bytes to store on a computer. Several levels of normalization of SMILES are recommended for systems that generate SMILES strings. Although these are not mandatory in any sense, they should be considered guidelines for software engineers creating SMILES systems.
4.2. No Normalization
The simplest "normalization" is no normalization. SMILES can be written in any form whatsoever, as long as they meet the rules for SMILES. Some examples of systems that might produce un-normalized SMILES are:
-
A system that enumerates combinatorial libraries using the rnum/dot-bond technique discussed above. SMILES produced by such a system will typically be a series of partial SMILES that are concatenated with dots into a complete molecule.
-
Simple pass-through "filters" that don’t have a full SMILES writer, but merely copy the input SMILES to the output. An example might be a molecular modeling program that reads SMILES to generates logP values, but has no capability to convert its molecular data structures back to a SMILES; instead it just copies its input SMILES to its output.
4.3. Standard Form
The "standard form" of a SMILES is designed to produce a compact SMILES, and one that is human readable (for smaller molecules).
In addition, a normalized SMILES has the important property that it matches itself as a SMARTS string. This is a very important feature of normalized SMILES in cheminformatics systems.
Note: In the example below, the "Wrong" SMILES examples are all valid SMILES, but are "wrong" in the sense that they are not the preferred form for standard normalization.
4.3.1. Atoms
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Write atoms in the "organic subset" as bare atomic symbols whenever possible. |
|
|
If the charge is |
|
|
If the hydrogen count is 1, leave off the digit. |
|
|
Always write the atom properties in the order: Chirality, hydrogen-count, charge. |
|
|
|
|
|
Represent hydrogens as a property of the heavy atom rather than as explicit atoms, unless other rules (e.g. |
4.3.2. Bonds
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Only write |
|
|
|
|
|
4.3.3. Cycles
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Don’t reuse ring-closure digits. |
|
|
Begin ring numbering with 1, not zero (or any other number) |
|
|
Avoid making a ring-closure on a double or triple bond. For the ring-closure digits, choose a single bond whenever possible. |
|
|
Avoid starting a ring system on an atom that is in two or more rings, such that two ring-closure bonds will be on the same atom. |
|
|
Use the simpler single-digit form for rnums less than 10. |
4.3.4. Starting Atom and Branches
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Start on a terminal atom if possible. |
|
|
Try to make "side chains" short; pick the longest chains as the "main branch" of the SMILES. |
|
|
Start on a heteroatom if possible. |
|
|
Only use dots for disconnected components. |
4.3.5. Aromaticity
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Write the aromatic form in preference to the Kekule form. |
4.3.6. Chirality
| Correct | Wrong | Normalization Rule |
|---|---|---|
|
|
Remove chiral markings for atoms that are not chiral. |
|
|
Remove cis/trans markings for double bonds that are not cis or trans. |
4.4. Canonical SMILES
A Canonical SMILES is one that follows the Standard Form above, and additionally, always writes the atoms and bonds of any particular molecule in the exact same order, regardless of the source of the molecule or its history in the computer. Here are a few examples of Canonical versus non-Canonical SMILES:
| Canonical SMILES | Non-canonical | Name |
|---|---|---|
|
|
ethanol |
|
||
|
|
phenol |
|
||
|
The primary use of Canonical SMILES is in cheminformatics systems. A molecule’s structure, when expressed as a canonical SMILES, will always yield the same SMILES string, which allows a chemical database system to:
-
Create a unique name (the SMILES) for each molecule in the system
-
Consolidate data about one molecule from a variety of sources into a single record
-
Given a molecule, find its record in the database
Canonical SMILES should not be considered a universal, global identifier (such as a permanent name that spans the WWW). Two systems that produces a canonical SMILES may use different rules in their code, or the same system may be improved or have bugs fixed as time passes, thus changing the SMILES it produces. A Canonical SMILES is primarily useful in a single database, or a system of related databases or information, in which all molecules were created using a single canonicalizer.
The rules (algorithms) by which the canonical ordering of the atoms in a SMILES are generated are quite complex, and beyond the scope of this document. There are many chemistry and mathematical graph-theory papers describing the canonical labeling of a graph, and writing a canonical SMILES string. See the Appendix for further information.
Those considering Canonical SMILES for a database system should also investigate InChI, a canonical naming system for chemicals that is an approved IUPAC naming convention.
4.5. SMILES Files
SMILES file consists of zero or more SMILES strings, one per line, optionally followed by at least one whitespace character (space or tab), and other data. There can be no leading whitespace before the SMILES string on a line. The optional whitespace character and data that follows it are not part of the SMILES specification, and interpretation of this data is up to applications that use the SMILES file. Each line of the file is terminated by either a singe LF character, or by a CR/LF pair of characters (commonly called the "Unix" and "Windows" line terminators, respectively). A SMILES parser must accept either line terminator. A blank line in the SMILES file, or a line that begins with a whitespace character, should be completely ignored by a SMILES parser.
5. Nonstandard Forms of SMILES
Several SMILES-generating systems are in use that either generate incorrect SMILES, or that interpreted some of the ambiguous features of the original SMILES specification in different ways. Although these SMILES are illegal according to this formal OpenSMILES specification, it is often useful to parse them, in order to make use of the information that accompanies these SMILES.
These "relaxed" SMILES rules should only be allowed when the user (presumably after thinking about the consequences) requests it. A SMILES parser that allows any or all of these "relaxed" rules must not do it by default. The user must specifically request these relaxed rules before a parser can accept such SMILES.
The following table lists "relaxed" rules that SMILES parsers may accept.
| Rule | Example | Interpreted as … | Details |
|---|---|---|---|
Extra parentheses |
|
|
Extra parentheses are ignored in places where there is no ambiguity as to the meaning. Note that the form |
|
|
||
|
|
||
Misplaced dots |
|
|
Two or more dot-bonds in a row are condensed into one. A leading or trailing dot-bond is ignored. Note that a dot that starts a branch is legal in strict SMILES; for example, |
|
|
||
|
|
||
Mismatched Ring Bonds |
|
|
Mismatched ring bonds are ignored. Note that this is almost always a bad idea. For example, |
Invalid Cis/Trans specification |
|
|
Mismatched or incomplete cis/trans bonds are ignored. |
|
|
||
|
|
||
Conflicting cis/trans specification |
|
|
Conflicting cis/trans bonds are ignored. (In this case, both the methyl and fluorine on the left are shown as trans to the methyl on the right, an impossible configuration.) |
D and T |
|
|
The symbols |
|
|
||
Lowercase as sp2 |
|
|
Lowercase letters are interpreted as sp2, even outside of ring systems. |
|
|
5.1. SMILES Flavors
It is an unfortunately common misconception that a Canonical SMILES does not contain stereochemistry (Minkiewicz et al. 2017) or alternatively that all SMILES must be canonical (Sykora and Leahy, 2008). SMILES flavors as described by Daylight are summarised below.
| Flavor | Atoms and Bonds Distinctly Ordered | Stereochemistry | Isotopes |
|---|---|---|---|
Canonical SMILES |
Y |
Y/N |
Y/N |
Arbitrary SMILES |
N |
Y/N |
Y/N |
Isomeric SMILES |
Y/N |
Y |
Y |
Unique SMILES |
Y |
N |
N |
Absolute SMILES |
Y |
Y |
Y |
Generic SMILES |
N |
N |
N |
These terms can be confusing and should be avoided due to conflicting definitions between vendors and toolkits. For example ChemAxon use the term Isomeric SMILES to mean a non-canonical SMILES with stereochemistry and isotopic information specified (see SMILES, ChemAxon Documentation). OEChem use the term Isomeric SMILES to mean a canonical SMILES with stereochemistry and isotopic information specified (see OECreateIsoSmiString), Absolute SMILES to mean a non-canonical SMILES with stereochemistry and isotopic information specified (see OECreateAbsSmiString), and Canonical SMILES to mean a canonical SMILES without stereochemistry and isotopic information specified (see OECreateCanSmiString).
In general the properties encoded in a SMILES can be chosen by a program to suit a particular purpose. You may have the option to independently include or omit stereochemistry, isotopes, or atom map/class in a generated SMILES. When referencing a particular SMILES flavor confusion can be avoided by including the toolkit, version, and options used.
6. Proposed Extensions
6.1. External R-Groups
Daylight proposed, and OpenEye actually implemented, an extension that
specifies bonds to external R-groups. An external R-group is specified
using ampersand '&' followed by a ring-closure specification (either a
digit, or '%' and two digits). However, unlike ring-closures, the bond is to
an external, unspecified R-group. Example: n1c(&1)c(&2)cccc1 - 2,3-substituted pyridine.
6.2. Polymers and Crystals
Daylight (Weininger) proposed, but never implemented, an extension for crystals and
polymers. Daylight also used the ampersand '&' character, (which may
conflict with the R-group proposal, above), but with the added rule that
if a number appears more than once, it creates a repeating unit.
| SMILES | Name |
|---|---|
|
polystyrene |
|
diamond |
|
graphite |
6.3. Atom-based Double Bond Configuration
The directional '/' and '\' marks for cis/trans bonds seem simple on
the surface but are problematic for complex systems. The issue is that
in conjugated systems one directional bond can be used in defining the
configuration of two double bonds. When assigning the directional bonds the
existing labels must be considered or rewritten. In a long series of
conjugated double bonds, changing the configuration of one bond can require
rewriting dozens of bond symbols.
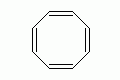
More importantly, there is a theoretical flaw with the use of '/' and
'\'. It is possible to write valid SMILES for the molecule
cyclooctatetraene by alternating
directional assignments for the cis configurations. However, as shown below attempting
to change one configuration is not possible. Reassigning the directional labels for
adjacent double bonds will not work as it reassignment propagates around the ring
and the conflict is not resolved.
Including directional labels to explicit hydrogen atoms is a possible resolution but does not follow standard-form and complicates the assignment procedure.
| Depiction | SMILES | Comment |
|---|---|---|
|
|
cyclooctatetraene |
Todo |
|
one bond changes two configurations |
The proposed syntax for double bond configurations uses the '@' and '@@' atom-based
specification. For example:
| Depiction | SMILES | Name |
|---|---|---|
|
|
trans-difluoroethene |
|
||
|
|
cis-difluoroethene |
|
Interpretation of '@' and '@@' follows the tetrahedral convention:
The atoms, as encountered in the SMILES string, are either in anticlockwise
'@' or clockwise '@@' order as viewed on the page. Since cis/trans
configurations are planar, they can also be "viewed from underneath the
page", which results in the two valid SMILES shown for each compound,
above.
As with the other atom-bases specifications one must consider the relative
position of implicit atoms. It is not always true that a trans form has
opposite "clock-ness" ('@','@@' or '@@','@'), and the cis form
has the same "clock-ness" ('@','@' or '@@','@@').
| Depiction | SMILES | Name |
|---|---|---|
|
|
trans-difluoroethene |
|
||
|
|
cis-difluoroethene |
|
Atom-based '@' and '@@' for the stereo-specification of double bonds does not
suffer from the theoretical flaw illustrated with cyclooctatetraene. The assignments
are not-shared and adjacent configurations do not need to be considered. This is more
flexible and and simplifies generation of canonical SMILES.
| Depiction | SMILES | Name |
|---|---|---|
|
|
cyclooctatetraene |
Note that the first stereo-specification carbon must be represented as '@' since the
'1' follows the H, whereas the rest of the carbons use '@@' to characterize the
cis configuration of each bond. Since this is a specification on the atom, rather than
the single bond, no conflict arises at the ring-closure bond.
6.4. Radical
This section needs considerable work. The following text is courtesy Chris Morley, who commented: "I guess the last paragraph doesn’t look too good in a formal specification. There are two reasons for the frailty: lack of proof that the radical and aromatic uses can always be unambigous (I doubt anybody has tried); and a known deficiency in the parser." However, it is a good starting point…
A single lowercase symbol is interpreted as a radical center. CCc is an alternative to CC[CH2] and
is the 1-propyl radical; CcC or C[CH]C is the 2-propyl radical, Co is the methoxy radical. An odd
number of adjacent lowercase symbols is a delocalized conjugated radical. So Cccccc is CC=CC=C[CH2]
or CC=C[CH]C=C or C[CH]C=CC=C Lowercase 'c' or 'n' can be used in a ring: C1cCCCC1 is the cyclohexyl
radical.
The use of the non-aromatic lowercase symbol is a shorted form with improved intelligibility that allows the use of implicit hydrogen in radicals. However it is intended only for simple unambiguous molecules and is not reliable when combined with aromatic atoms.
6.5. Twisted SMILES
An interesting extension that specifies conformational information via bond dihedral angles and bond lengths was proposed by McLeod and Peters:
7. APPENDIX 1: References and Citations
7.2. Documentation
Daylight
The most-referenced definition of SMILES on the web.
Article by Roger Sayle about converting PDB files to SMILES with thorough treatment of aromaticity.
OpenEye
OpenBabel
Wikipedia
7.3. Toolkits
OpenBabel
The Chemistry Development Kit
Marvin
RDKit
Frowns
PerlMol.org
InChI
7.4. Some Key Scientific Papers
-
David Weininger, SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules, Journal of Chemical Information and Computer Sciences, 1988, 28:31-36.
-
David Weininger, Arthur Weininger, and Joseph L Weininger, SMILES 2. Algorithm for Generation of Unique SMILES Notation Journal of Chemical Information and Computer Sciences, 1989, 29:97-101.
-
Morgan’s original canonicalization paper: Morgan, H.L. J.Chem.Doc. 1965, 5, 107
-
G.M. Downs et al, Review of Ring Perception Algorithms for Chemical Graphs, J. Chem. Inf. Comput. Cci. 1989, 29, 172-187
-
R.Balducci and R, Pearlman, Novel Algorithms for the Rapid Perception of a Unique Optimal Set of Rings, J. Am. Chem. Soc. (date?)
7.5. Molecule Editors that can produce SMILES
-
JME
-
CACTVS
-
ISISDraw?
-
ChemDraw
-
ACD/ChemSketch
8. Revision History
| Revision | Date | Description | Name |
|---|---|---|---|
1.0 |
2007-11-13 |
Draft |
Craig A. James |
1.0 |
2012-09-29 |
Reformatting |
Tim Vandermeersch |
1.0 |
2012-09-29 |
Corrections |
Andrew Dalke & Tim Vandermeersch |
1.0 |
2012-11-17 |
SP, TB and OH stereochemistry |
Tim Vandermeersch |
1.0 |
2013-09-06 |
Corrections |
Richard Apodaca |
1.0 |
2013-09-17 |
Corrections |
John May |